Czym jest dystrofia mięśniowa Duchenne’a?
Dystrofia mięśniowa Duchenne’a (DMD) stanowi najczęściej występujące zaburzenie neuromięśniowe wieku dziecięcego, dotykające w przybliżeniu 1 na 5000 nowo narodzonych chłopców1. DMD wywoływana jest przez mutacje w genie dystrofiny, największym genie w ludzkim genomie. Większość chłopców z DMD posiada mutacje prowadzące do braku lub poważnej redukcji ilości białka o nazwie dystrofina.
Kliniczne cechy DMD obejmują opóźnienie kamieni milowych rozwoju, trudności ze skakaniem i bieganiem, utratę umiejętności chodzenia w okresie dojrzewania oraz postępującą niewydolność krążeniową i oddechową2.
Objawy kliniczne stają się widoczne w wieku od 2 do 4 lat, ale na całym świecie donosi się o opóźnieniach w rozpoznawaniu chorych chłopców, a średnia postawienia diagnozy wynosi 4,3 roku w Wielkiej Brytanii3 i 4,9 roku w Stanach Zjednoczonych4,5. Choć fenotyp jest stosunkowo jednorodny, zgłaszano pewne związki między genotypem a fenotypem (np. podwojenie eksonu 44 lub 45 zwykle skutkuje łagodniejszym fenotypem)6-8.
Pierwsze zalecenia na temat opieki zdrowotnej przy dystrofii mięśniowej Duchenne’a opublikowano w 2010 r., a niedawno je uaktualniono9,10,2. Wdrożenie opieki znacząco zmieniło naturalny przebieg DMD – chłopcy obecnie dłużej zachowują zdolność chodzenia, później dochodzi u nich do niewydolności krążeniowo-oddechowej, rzadziej potrzebują operacji kręgosłupa i żyją dłużej11,12.
Choć w ostatnich latach szereg publikacji opisywał miary rezultatów w DMD13, większość z tych badań dotyczyła chłopców leczonych przy pomocy kortykosteroidów i przechodzących rozmaite interwencje fizjoterapeutyczne.
Genotyp, leczenie sterydami i fizjoterapia mogły wpływać na fenotyp i wyniki kliniczne7,14,15,11. W związku z tym nie opisano dotąd fenotypu populacji młodych pacjentów z DMD nie poddawanych leczeniu.
Skala North Star Ambulatory Assessment (NSAA – badanie zdolności do chodzenia) została uznana jako klinicznie znacząca przez odpowiednie instytucje i obecnie jest regularnie stosowana w badaniach klinicznych nad DMD jako kliniczny punkt końcowy w pomiarach skuteczności leków16,17. Wykazano, że NSAA jest skalą wiarygodną i nadającą się psychometrycznie do stosowania u pacjentów w różnym wieku18-20; nie została ona jednak oceniona w populacji złożonej wyłącznie z chłopców nie przyjmujących steroidów.
Jedynie ograniczone dane opisują miary rezultatów używane w próbach młodych pacjentów nie stosujących steroidów oraz różnice między nimi a zdrowymi chłopcami w tym samym wieku21. Ponadto nie jest znana wiarygodność takich miar rezultatu u młodych pacjentów z DMD. Scharakteryzowanie sprawności ruchowej przy użyciu testów czynnościowych i w mierzonym czasie u młodych chłopców z DMD nie stosujących steroidów mogłoby pomóc w zidentyfikowaniu właściwych miar rezultatów dla tej populacji, co z kolei mogłoby pomóc w procesie diagnostycznym, wspieraniu wczesnego rozpoznawania choroby i projektowaniu badań klinicznych z udziałem młodszych pacjentów.
Badanie FOR-DMD jest randomizowanym, podwójnie zaślepionym badaniem komparatywnym nad korzyściami i efektami ubocznymi trzech najczęściej przepisywanych schematów stosowania kortykosteroidów w DMD. Jego celem jest zidentyfikowanie najlepszego schematu stosowania tych leków, jeśli chodzi o równowagę korzyści i ryzyka, i ustalenie standardów opieki w tej populacji22.
Dane początkowe z badania FOR-DMD dostarczają szerokiego opisu sprawności ruchowej chłopców w wieku od 4 do 7 lat nie stosujących sterydów. Dane te poszerzą wiedzę na temat wczesnych objawów choroby i pomogą w kształtowaniu kryteriów włączania uczestników do innych badań terapeutycznych.
Metody badania schematów postępowania w dystrofii mięśniowej Duchenne’a (FOR-DMD)
Rekrutacja do badania FOR-DMD ograniczała się do chłopców z genetycznie potwierdzonym rozpoznaniem DMD i klinicznymi objawami choroby. Wiek podczas skriningu mieścił się w przedziale od 4 do 8 lat. Warunkami włączenia do badania były między innymi: zdolność do samodzielnego wstania z podłogi; gotowość/zdolność do stawiania się na umówione wizyty, stosowania się do planu podawania leków i procedur badawczych oraz zdolność do uzyskiwania powtarzalnych wyników pomiarów natężonej pojemności życiowej (FVC – forced vital capacity), czyli z różnicą między najlepszymi wynikami FCV w dwóch różnych próbach podjętych podczas skriningu mniejszą od 20%. Chłopcy, którzy przeszli wcześniej leczenie kortykosteroidami lub innymi lekami immunosupresyjnymi, a także przyjmujący w czasie rekrutacji lub w ciągu 3 miesięcy przed momentem początkowym jakikolwiek lek badawczy byli wykluczani z badania. Dostępny jest opublikowany pełny opis projektu badania FOR-DMD22.
Podczas skriningu i w momencie początkowym zebrano dane na temat sprawności ruchowej przed leczeniem. Protokół zezwalał na okno między skriningiem (data pozyskania zgody) a momentem wyjściowym (data podania pierwszej dawki badanego leku) o czasie trwania do 3 miesięcy (90 dni).
Podczas skriningu i w momencie początkowym oceniano następujące miary rezultatów:
- funkcję oddechową, mierzoną przez FVC w pozycji siedzącej23,22;
- sprawność czynnościową, w tym badanie North Star Ambulatory Assessment (NSAA) obejmujące zdolność wstawania z podłogi i mierzącą czas wykonywania tej czynności oraz zdolność do przejścia/przebiegnięcia 10 m i czas wykonania tego zadania19,18, a także
- dystans pokonany podczas sześciominutowego testu marszowego (6MWT – 6-minute walk test)24,25.
Wszystkie oceny sprawności ruchowej przeprowadzali fizjoterapeuci z doświadczeniem w chorobach neuromięśniowych, którzy przeszli standaryzowane i rygorystyczne szkolenie na temat protokołu badania FOR-DMD. Jakość danych była kontrolowana poprzez weryfikację wideo online przeprowadzaną przez dwóch specjalistów zakresu fizjoterapii neuromięśniowej w celu sprawdzenia zgodności z procedurami.
Wyniki badania FOR-DMD
Dane kliniczne pacjentów zakwalifikowanych do badania FOR-DMD
W wizycie początkowej udział wzięło 196 chłopców zakwalifikowanych do badania. Dane kliniczne pacjentów zebrane na wizycie początkowej podsumowuje tabela 1. Dystrybucja typów mutacji u uczestników była zgodna z wcześniejszymi dużymi badaniami kohortowymi26.
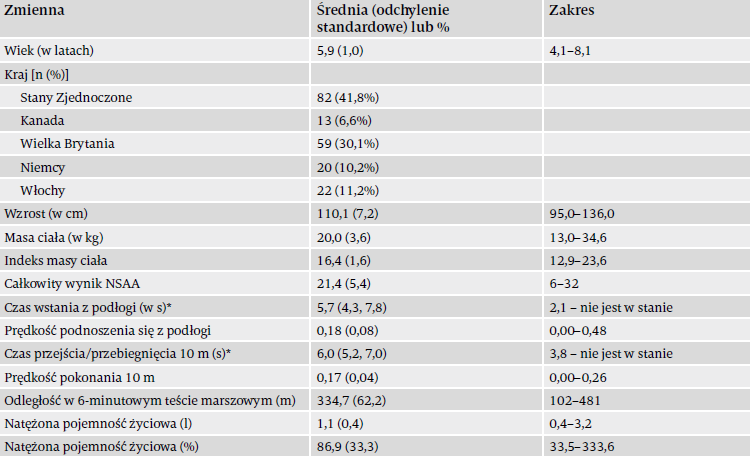
Związek między wynikami funkcjonalnymi a wiekiem w FOR-DMD
Znaczące statystycznie były następujące porównania między grupami:
- wynik całkowity NSAA – 6 lat vs 4 lata (p=0,02), 7 lat vs 6 lat (p=0,045);
- prędkość wstawania z podłogi – 7 lat vs 4 lata (p=0,049), 7 lat vs 5 lat (p < 0,0001), 7 lat vs 6 lat (p=0,0005);
- prędkość testu 10 metrów – 5 lat vs 4 lata (p=0,04), 6 lat vs 4 lata (p=0,03), 6 lat vs 5 lat (p=0,004), 7 lat vs 6 lat (p=0,004);
- dystans w 6-minutowym teście marszowym – 6 lat vs 4 lata (p=0,006);
- natężona pojemność życiowa (FVC) – 6 lat vs 4 lata (p=0,001), 6 lat vs 5 lat (p=0,04), 7 lat vs 4 lata ( p < 0,0001), 7 lat vs 5 lat (p=0,0002).
Stwierdzono umiarkowane do silnych korelacje między testami czynnościowymi wykonywanymi w mierzonym czasie a całkowitym wynikiem NSAA. Korelacje między tymi pomiarami a FVC były natomiast bardzo słabe.
Rzetelność wyniku wg metody powtórnego pomiaru (test-retest) uzyskanego w FOR-DMD
Na 196 chłopców biorących udział w badaniu dysponowano danymi:
- 152 (6-minutowy test marszowy),
- 163 (całkowity wynik NSAA w tym testy na czas) oraz
- 149 (wartości FVC) uczestników w celu oszacowania rzetelności.
Rzetelność ta okazała się ogólnie akceptowalna we wszystkich grupach wiekowych, jednak była dość niska dla 6-minutowego testu marszowego u chłopców poniżej 5. roku życia.
Omówienie badania FOR-DMD
Dane początkowe z badania FOR-DMD zapewniają pierwszy szczegółowy opis czynnościowych miar rezultatów i testów wykonywanych w mierzonym czasie rutynowo stosowanych w praktyce klinicznej i w badaniach klinicznych w dużej kohorcie pacjentów z DMD przed podjęciem leczenia (czyli nie przyjmujących steroidów).
Co ważne, wykazano, że wszystkie testy funkcjonalne (prędkość wstawania z podłogi oraz prędkość pokonania dystansu 10 m, całkowity wynik NSAA oraz dystans 6MWT) mają akceptowalną rzetelność (porównanie wizyty skriningowej z wizytą początkową), bez względu na czas między ocenami (mniej niż 30 dni albo w przedziale od 31 do 90 dni). Podczas analizy według grup wiekowych u pacjentów w wieku co najmniej 5 lat wykazano akceptowalną rzetelność wszystkich testów, a u pacjentów poniżej 5 lat jedynie 6MWT charakteryzował się słabą rzetelnością. Największą szacowaną rzetelność miał całkowity wynik NSAA. Wyniki te są w zgodzie z ustaleniami z wcześniejszych publikacji27.

Jak (…) wykazano na kohortach przechodzących steroidoterapię, sam wiek nie jest dobrym predyktorem ciężkości choroby.
Do projektu tego badania włączono wiarygodny element, jakim jest pomiar FVC, przy czym dwudziestu trzem chłopcom nie udało się przejść skriningu ze względu na niemożność uzyskania spójnych wyników FVC podczas wizyty skriningowej. Dla tych, którzy byli w stanie wykonać wiarygodny pomiar FVC na tamtym etapie, wykazano rzetelność tego pomiaru (porównanie wyniku podczas skriningu z wynikiem podczas wizyty początkowej) bez względu na czas, jaki upłynął między tymi ocenami i we wszystkich grupach wiekowych. Oczywiste także było, że średnia FVC u chłopców w wieku 7 lat statystycznie różniła się od średniej u chłopców w wieku 4 i 5 lat, a także średni wynik FVC w grupie 6-latków statystycznie różnił się od wyników uzyskanych u 4- i 5- latków. Słaba korelacja między FVC a testami czynnościowymi może wynikać z wyraźnej różnicy między konstrukcją badania FVC a pozostałych pomiarów oraz z faktu, że podczas rozwoju chłopców, jaki zachodzi między wiekiem 4 a 7 lat, spodziewany jest także wzrost wyniku FVC (tabela 2).
Ewentualna stronniczość w ocenianiu rzetelności miar rezultatów może wynikać z faktu, że chęć i zdolność do poddania się pomiarom związanym z tym badaniem stanowiła jedno z kryteriów włączających, przez co doszło do wykluczenia chłopców z poważnymi trudnościami z uczeniem się i problemami behawioralnymi. Choć może to stanowić stronniczość, takie kryteria włączające są powszechnie stosowane w klinicznych badaniach nad DMD.
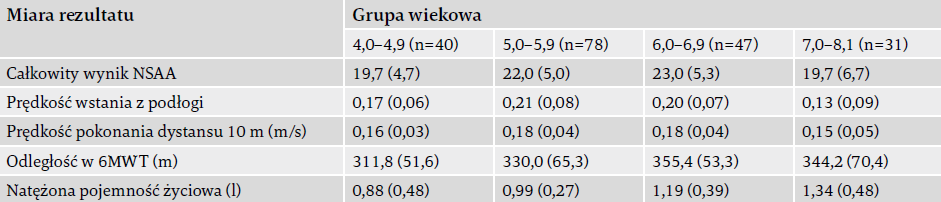
Średnia prędkości wstawania z podłogi uzyskana przez chłopców w wieku 7 lat była statystycznie różna od średniej chłopców w wieku 4, 5 i 6 lat. Prędkość pokonania dystansu 10 m także była różna w poszczególnych grupach wiekowych (5 vs 4, 6 vs 4, 6 vs 5 i 7 vs 6 lat), co sugeruje, że obie te miary rezultatów wydają się odzwierciedlać zyski rozwojowe i progresję choroby u pacjentów bez steroidoterapii.
Dane z niniejszego badania są spójne z danymi opublikowanymi na temat chłopców leczonych steroidami28, które wykazywały dobry związek między całkowitym wynikiem NSAA a dystansem 6MWT29 oraz NSAA a testami w mierzonym czasie. Stosunkowo niską korelację zaobserwowano w próbie niniejszego badania między prędkością wstawania z podłogi a dystansem 6MWT – podobne ustalenie zgłaszali Mazzone i wsp. u chłopców podczas terapii steroidami30. Przyczyna tego może mieć związek z typem aktywności mięśni biorących udział w każdym z testów oraz dosyć niewielkiej siły mięśni wymaganej do chodzenia, gdy już pacjent wstał i się porusza.
Fenotyp młodej populacji DMD nie stosującej steroidoterapii nie został dotąd dobrze opisany. Niniejsza przekrojowa próba poprawi nasze zrozumienie przejawiania się DMD przed wdrożeniem terapii steroidami, a dalsze prace mogą skupiać się na ocenie związków między genotypem a fenotypem przy braku znaczących czynników mylących, takich jak steroidoterapia i schemat jej stosowania. Ponadto dostarcza ona wyjściowych danych do planowanych badań klinicznych w populacjach nie stosujących steroidów, w których funkcjonalne kryteria włączające i wyłączające mają kluczowe znaczenie podczas rekrutacji. sam wiek nie jest dobrym predyktorem ciężkości choroby13.
W badaniu niniejszym z udziałem młodych chłopców nie stosujących steroidów, mimo że zaobserwowano znaczące różnice w miarach funkcjonalnej sprawności między grupami wiekowymi, różnice te nie są wystarczająco wyraźne, by definiować zdolności funkcjonalne, a wiek nie powinien być stosowany w izolacji jako kryterium włączające do badań klinicznych.
Wnioski z badania FOR-DMD
Badania kliniczne nad DMD często są skomplikowane i wiążą się z intensywnymi i rygorystycznymi protokołami. Harmonogramy wizyt, zwłaszcza na etapie skriningu i momentu początkowego, mają znaczący wpływ na obciążenie związane z badaniem dla rodzin pacjentów i dla klinicystów, którzy muszą zorganizować wymagane badania w ramach okna czasowego, na jakie zezwala protokół. Badanie FOR-DMD potwierdza, że u młodych chłopców z DMD można uzyskać wiarygodne pomiary rezultatów, a odstęp czasowy między badaniami nie przekraczający 90 dni nie wpływa na ich rzetelność. Tylko u chłopców poniżej 5. roku życia wyniki 6MWT mogą być mniej wiarygodne niż te uzyskane u pacjentów starszych. Ustalenie to może mieć wpływ także na kryteria włączające. W niniejszym badaniu dużą rolę odegrało odpowiednie przeszkolenie osób przeprowadzających oceny kliniczne oraz wdrożenie systemów kontroli jakości. Te obserwacje należy wziąć pod uwagę podczas planowania badań klinicznych. Testy funkcjonalne w mierzonym czasie wstawania z podłogi oraz pokonania dystansu 10 m wykazały statystycznie znaczące różnice między grupami wiekowymi i stanowią wiarygodne, nieczasochłonne testy do wykorzystywania nie tylko w badaniach, ale także w praktyce klinicznej.
Źródło: Neuromuscular Disorders. 2022; 32: 460-467 ©2022 The Authors
Adaptacja: Katarzyna Bogiel Na podstawie licencji CC BY (http://creativecommons.org/licenses/by/4.0/)
- Mendell JR, Shilling C, Leslie ND, Flanigan KM, Al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 2012;71:304–13. doi: 10. 1002/ ana.23528 .
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Colvin MK, et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol 2018;17:445–55. doi: 10.1016/S1474- 4422(18)30026- 7 .
- van Ruiten HJA, Straub V, Bushby K, Guglieri M. Improving recognition of Duchenne muscular dystrophy: a retrospective case note review. Arch Dis Child 2014;99:1074–7. doi: 10.1136/ archdischild- 2014- 306366 .
- Ciafaloni E, Fox DJ, Pandya S, Westfield CP, Puzhankara S, Romitti PA, et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the muscular dystrophy surveillance, tracking, and research network (MD STARnet). J Pediatr 2009. doi: 10.1016/j.jpeds.2009.02.007 .
- Soim A, Smith MG, Kwon JM, Mann JR, Thomas S, Ciafaloni E. Is there a delay in diagnosis of Duchenne muscular dystrophy among preterm-born males? J Child Neurol 2018. doi: 10.1177/ 0883073818773029.
- Ricotti V, Ridout DA, Pane M, Main M, Mayhew A, Mercuri E, et al. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J Neurol Neurosurg Psychiatry 2016. doi: 10.1136/jnnp- 2014- 309405 .
- Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol 2011;258:1610–23. doi: 10. 1007/s00415- 011- 5979- z.
- Davidson ZE, Kornberg AJ, Ryan MM, Sinclair K, Cairns A, Walker KZ, et al. Deletions in the dystrophin gene predict loss of ambulation before 10 years of age in boys with Duchenne muscular dystrophy. Neuromuscul Disord 2012;22:835. doi: 10.1016/j.nmd.2012. 06.111 .
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018;17:211–12. doi: 10.1016/S1474- 4422(18)30024- 3 .
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018;17:347–61. doi: 10.1016/ S1474- 4422(18)30025- 5 .
- McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, et al. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet 2018. doi: 10.1016/S0140-6736(17) 32160-8 .
- Bushby K, Muntoni F, Urtizberea A, Hughes R, Griggs R. Report on the 124th ENMC international workshop. Treatment of Duchenne muscular dystrophy; defining the gold standards of management in the use of corticosteroids 2-4 April 2004, Naarden, The Netherlands. Neuromuscul Disord 2004;14:526–34. doi: 10.1016/j.nmd.2004.05.006 .
- Muntoni F, Domingos J, Manzur AY, Mayhew A, Guglieri M, Sajeev G, et al. Categorising trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS ONE 2019. doi: 10.1371/journal.pone.0221097 .
- Ricotti V , Ridout DA , Pane M , Main M , Mayhew A , Mercuri E , et al. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J Neurol Neurosurg Psychiatry 2016;87:149–55 .
- Mercuri E, McDonald C, Mayhew A, Florence J, Mazzone E, Bianco F, et al. International workshop on assessment of upper limb function in Duchenne Muscular Dystrophy. Rome, 15-16 February 2012. Neuromuscul Disord 2012;22:1025–8. doi: 10.1016/j.nmd.2012.06.006 .
- Bushby K., Kirschner J., Luo X., Elfring G., Kroger H., Riebling P., et al. Results of North Star Ambulatory Assessments (NSAA) in the phase 3 ataluren confirmatory trial in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD) (I15.008). 2016.
- McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double- -blind, placebo-controlled, phase 3 trial. Lancet 2017. doi: 10. 1016/S0140- 6736(17)31611- 2.
- Mayhew A , Cano S , Scott E , Eagle M . Moving towards meaningful measurement: rasch analysis of the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Med Child 2011 .
- Scott E, Eagle M, Mayhew A, Freeman J, Main M, Sheehan J, et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int 2012;17:101–9. doi: 10.1002/pri.520.
- Hobart J, Cano S. Improving the evaluation of therapeutic interventions in multiple sclerosis: the role of new psychometric methods. Heal Technol Assess 2009;13:1–177 iii, ix–x. doi: 10.3310/hta13120.
- Goemans N, Klingels K, van den Hauwe M, Boons S, Verstraete L, Peeters C, et al. Six-minute walk test: reference values and prediction equation in healthy boys aged 5 to12 years. PLoS ONE 2013;8:e84120. doi: 10.1371/journal.pone.0084120.
- Guglieri M, Bushby K, McDermott MP, Hart KA, Tawil R, Martens WB, et al. Developing standardized corticosteroid treatment for Duchenne muscular dystrophy. Contemp Clin Trials 2017;58:34–9. doi: 10.1016/j.cct.2017.04.008 .
- Griggs RC. The use of pulmonary function testing as a quantitative measurement for therapeutic trials. Muscle Nerve 1990;13 Suppl:S30- 4.. doi: 10.1002/mus.880131310.
- McDonald CM, Henricson EK, Han JJ, Abresch RT, Nicorici A, Elfring GL, et al. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve 2010;41:500–10. doi: 10. 1002/mus.21544.
- Crapo RO, Casaburi R, Coates AL, Enright PL, MacIntyre NR, McKay RT, et al. ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002. doi: 10.1164/ajrccm.166.1.at1102.
- Schiava M, Amos R, VanRuiten H, McDermott MP, Martens WB, Gregory S, et al. Clinical and genetic characteristics in young, glucocorticoid-naive boys with Duchenne muscular dystrophy. Neurology 2022;98:e390–401. doi: 10.1212/WNL.0000000000013122.
- Mazzone ES, Messina S, Vasco G, Main M, Eagle M, D’Amico A, et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromuscul Disord 2009;19:458–61. doi: 10.1016/j. nmd.2009. 06.368 .
- McDonald CM, Henricson EK, Abresch RT, Florence J, Eagle M, Gappmaier E, et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve 2013;48:357–68. doi: 10.1002/mus.23905 .
- Pane M, Mazzone ES, Sivo S, Sormani MP, Messina S, D’Amico A, et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS ONE 2014;9. doi: 10. 1371/ journal.pone.0108205 .
- Mazzone E, Martinelli D, Berardinelli A, Messina S, D’Amico A, Vasco G, et al. North Star Ambulatory Assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord 2010;20:712–16. doi: 10.1016/j.nmd.2010.06.014. 467